The server let users to structurally annotate the mutations with using the StructMAn
Go ahead & try it out! You can easily just try out the server and explore the possibilities.
If you don't know what input is expected, you can check out the examples under the documentation page. If you have any questions, check out the FAQ.
StructMan is freely available to all users without any login requirement.
-
What is StructMAn
The Structural Mutation Annotation (StructMAn) software provides annotation of non-synonymous single-nucleotide variants (nsSNVs) in the context of the structural properties of the resulting amino acid changes in the corresponding proteins. Its rationale is that if a mutation is located on an interaction interface between a protein and another protein, DNA, RNA or a small molecule, it is likely to interfere with this interaction, and mutations location in the protein core are likely to influence its stability. StructMAn was first published in Nucl Acids Res, 2016 where we showed that such structural annotation correlate well with established tools for predicting damaging effect of missense mutations. Later we have applied StructMAn to a large collection of disease-associated and neutral mutations and discovered distinct trends of their spatial distribution.
-
Which input is expected?
The StructMAn server accepts the following inputs:
- Amino acid sequence (FASTA format)
- (human) Ensemble transcript identifier
- SMLF (simple mutation list format) file, you can find information here: SMLF - Structman docs
or example SMLF file here: example SMLF file
-
What is protein size limit?
- The StructMAn server accepts maximum 10 proteins as fasta input
- For larger proteins, you can try to install StructMAn on your local PC/server, you can find more information on our GitHub page
-
Which proteins are supported?
- The StructMAn server supports all the proteins
Last update: 27.05.2026
News
- - Inclusion of the AlphaFold DB collaboration datasets
- We updated our structure database with the newest additions to the Alphafold Structure database in the form of the collaboration datasets that include high quality structure predictions for bacterial and viral proteins. (AlphaFold Database welcomes community datasets | EMBL-EBI)
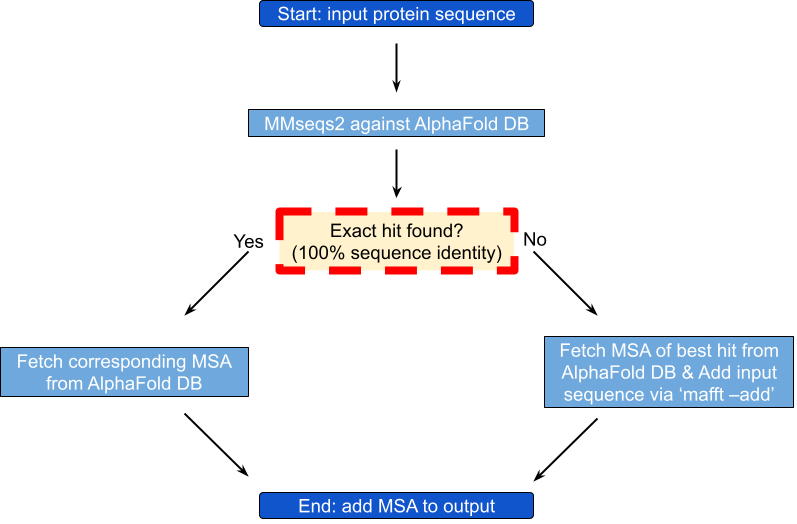
- - Addition of MSAs to StructMAn outputs
- We now provide MSAs for any protein sequence based on the MSAs that got recently added to the AlphaFold DB (EMBL-EBI and Google DeepMind renew partnership and release update to AlphaFold Database)
- A sequence similarity search for the given protein is performed against the Alphafold Protein Structure Database. If the search results include an exact hit (100% sequence identity) the corresponding MSA is fetched from the Alphafold Protein Structure Database and added to our output. Otherwise, the MSA from the best hit is retrieved and the given sequence is added to the MSA using “mafft –add” (https://mafft.cbrc.jp/alignment/server/add.html) and the resulting MSA is added to our output.
- - Addition of predicted complex structures
- Leveraging the results from the recent work from Zhang et al. (Predicting protein-protein interactions in the human proteome | Science), we integrated the predicted complex structures in our structure database, enabling them for annotation by StructMAn, further increasing the capability of StructMAn Web to identify Protein-Protein-Interactions.
- This prepares StructMAn for the next upcoming Alphafold DB update (Millions of protein complexes added to AlphaFold Database shed light on how proteins interact | EMBL) that will add predicted complex structures to the AlphaFold DB, which in turn we will add to our structure database.
- - Update of StructMAn backend to version 2.3.0